

THIẾT KẾ CÁC DẪN XUẤT UREA CÓ KHẢ NĂNG CHỐNG OXY HOÁ CHO SẢN PHẨM DẦU MỎ
MỞ ĐẦU
1. Lý do chọn đề tài
Các hợp chất dẫn xuất từ urea đóng vai trò rất quan trọng trong đời sống và thu hút quan tâm nghiên cứu.
Hoá học tính toán với trợ giúp của phần mềm cho kết quả chính xác cao.
Oxy hoá trong các sản phẩm dầu mỏ dẫn đến ăn mòn động cơ, giảm độ bền vật liệu …
Các hợp chất được tổng hợp từ các tiền chất urea là những chất chống oxy hóa tốt.
Do đó chúng tôi đề xuất nghiên cứu đề tài “THIẾT KẾ CÁC DẪN XUẤT UREA CÓ KHẢ NĂNG CHỐNG OXY HOÁ CHO SẢN PHẨM DẦU MỎ”.
2. Tổng quan tài liệu và tình hình nghiên cứu đề tài
3. Mục đích và nội dung nghiên cứu
3.1. Mục tiêu
Thiết kế và nghiên cứu các dẫn xuất mới từ urea có hoạt tính chống oxy hóa tốt.
3.2. Nội dung nghiên cứu
- Tìm kiếm các hệ chất dẫn xuất urea mới có khả năng chống oxy hóa;
- Tính toán các thông số nhiệt động học liên quan như BDE, PA, IE, PDE…;
- Đánh giá hoạt tính chống oxy hóa dựa trên các thông số nhiệt động học và bề mặt thế năng phản ứng;
- Xây dựng mối quan hệ và phân tích sự tương quan giữa các thông số nhiệt động và thông số cấu trúc, tìm ra sự liên hệ giữa các thông số;
- Thiết kế các hợp chất mới từ dẫn xuất urea có hoạt tính chống oxy hóa cao;
- Thực hiện một vài thí nghiệm kiểm.
4. Đối tượng và phạm vi nghiên cứu
4.1. Đối tượng nghiên cứu
Nghiên cứu hệ chất như Hình 1 và so sánh với hệ chất ở Hình 2.
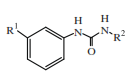
Hình 1. Hệ chất nghiên cứu ban đầu
R1 = H, halogen, alkyl, phenyl… và R2: H, phenyl…
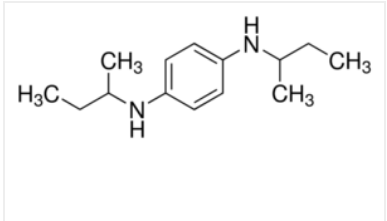
N-N’-di-sec-butyl-p-phenylenediamin
Đề tài chỉ tập trung vào nghiên cứu một cách hệ thống hoạt tính chống oxy hoá của các dẫn xuất bất đối xứng của urea như Hình 3 thông qua các thông số nhiệt động, đồng thời làm rõ cơ chế và phương thức chống oxy hoá đặc trưng dựa trên cơ chế chuyển nguyên tử hydro (HAT).
Hình 3. Cấu trúc của 1-(3-methylbenzyl)-3-phenylurea cùng dẫn xuất thiourea và selenourea analogues (X= O, S and Se).
3.2. Phạm vi nghiên cứu
- Tính toán các thông số nhiệt động liên quan BDE, IE, PA… và thông số cấu trúc khác; phân tích sự ảnh hưởng của nhóm thế đến sự thay đổi các thông số trên. Từ đó tối ưu hóa cấu trúc các hợp chất của hệ nghiên cứu.
- Đề xuất và thiết kế các hợp chất có hoạt tính chống oxy hóa tốt.
- Đánh giá và so sánh khả năng chống oxy hóa của N-N’-di-sec-butyl-p-phenylenediamin (phụ gia chống oxy hóa cho xăng) với hệ chất nghiên cứu.
Đề tài tập trung vào việc nghiên cứu một cách có hệ thống hoạt tính chống oxy hoá của các dẫn xuất Urea bằng cách thay thế các nguyên tử Nitơ bằng nhóm benzyl và phenyl nhằm làm rõ đặc trưng của cơ chế và phương thức chống oxy hoá dựa trên quá trình dịch chuyển nguyên tử Hydro (HAT). Qua đó, giải quyết các vấn đề:
– Quan sát và đánh giá cấu cấu trúc PU;
– Xem xét quá trình dịch chuyển nguyên tử Hydro xảy ra tại liên kết C-H hay N-H trong hợp chất urea;
– Ảnh hưởng của nhóm thế methyl tại vị trí ortho và para của vòng benzyl;
– Ảnh hưởng của oxy đến khả năng chống oxy hoá của PU;
– Thiết kế các hợp chất 1-(3-methylbenzyl)-3-phenylurea (PTU) và 1-(3-methylbenzyl)-3-phenylselenourea (PSU) làm cơ sở để tính toán và so sánh.

5. Phương pháp nghiên cứu
- Tổng quan lý thuyết, các phương pháp hóa tính toán của hoá học lượng tử; tổng quan các nguồn tài liệu;
- Ứng dụng phần mềm Gaussian 09, Gaussview 5;
- Áp dụng thuyết phiếm hàm mật độ (DFT) trong hoá học lượng tử;
- Ứng dụng ba cơ chế của phản ứng (HAT, SET-PT, SPLET), các nhiệt động (BDE, IE, PDE, PA, ETE…) để làm rõ quá trình ức chế và dập tắt gốc tự do;
- Nghiên cứu và tính toán bề mặt thế năng (PES) thông qua hợp chất trung gian và trạng thái chuyển tiếp;
- Sử dụng qui trình tính toán ROB3LYP/6-311++G(2df,2p)//B3LYP/6-311G(d,p) để áp dụng trong phần mềm Gaussian 09;
- Tính toán động học bằng cách sử dụng phần mềm Eyringpy.
6. Ý nghĩa khoa học và thực tiễn đề tài
Kết quả đạt được sẽ phục vụ cho cơ sở lý thuyết trong việc nghiên cứu các hợp chất hóa học; tìm ra được nhiều chất chống oxy hóa phù hợp để ứng dụng vào thực tế. Do đó, đề tài vừa có ý nghĩa khoa học, vừa có ý nghĩa thực tiễn, đặc biệt trong lĩnh vực dầu mỏ và polymer.
Chương 1 – TỔNG QUAN HỆ CHẤT NGHIÊN CỨU
Tổng quan về chất oxy hóa và chất chống oxy hóa
Việc nghiên cứu các hợp chất oxy hoá và các hợp chất chống oxy hoá có vai trò rất quan trọng trong thực tiễn.
Trong hoá sinh, nghiên cứu được ứng dụng cho quá trình chống oxy hoá trong thực phẩm, vitamin, chống lão hoá cơ thể người…
Trong công nghiệp, chất chống oxy hoá được dùng cho nhiên liệu, dầu mỡ bôi trơn, sơn chống ăn mòn…
Gốc tự do
Gốc tự do được hình thành bởi một phân tử có một điện tử độc thân chưa được ghép đôi, chưa tạo thành cặp. Gốc tự do liên tục được sinh ra và phát triển theo chuỗi phản ứng liên hoàn.
1.1.2. Chất chống oxy hóa
Là các hợp chất làm chậm hoặc dập tắt quá trình oxy hoá. Chất chống oxy hoá thường được bổ sung vào quá trình phản ứng để làm chậm hay dừng quá trình oxy hoá.
1.1.3. Cơ chế chống oxy hóa
Quá trình chống oxy hoá có thể mô tả và diễn giải theo ba cơ chế: HAT, SET-PT, SPLET
1.1.3.1. Cơ chế hat (hydrogen atomic transfer)
UH + ROO• → U• + ROOH
Gốc U• tạo thành tiếp tục ứng với các gốc tự do khác để tạo thành các sản phẩm khác và không sinh ra thêm gốc tự do nào. Hiệu quả của cơ chế được quyết định bởi enthalpy phân cắt đồng ly (BDE) của liên kết N-H, xác định theo phương trình:
BDE(U-H) = H(U•) + H(H•) – H(UH)
1.1.3.2. Cơ chế set-pt (single electron transfer−proton transfer)
Cơ chế SET-PT thực hiện qua hai quá trình sau:
UH + ROO• → UH+• + ROO• (1) → U• + ROOH (2)
Đánh giá khả năng oxy hoá của một hợp chất theo cơ chế SET-PT nhờ vào các giá trị IE và PDE, các giá trị này được xác định theo phương trình:
IE= Hf(UH+•) + Hf(e–) – H (UH)
PDE= Hf(UN•) + Hf(H+) – H(UH+•)
1.1.3.3. Cơ chế SPLET (Sequential Proton Loss Electron Transfer)
Cơ chế SPLET gồm hai bước:
– Mất proton H+: UH → U− + H+
Bước này được đặc trưng bởi thông số ái lực proton – PA
– Dịch chuyển electron: U− + ROO• → U• + ROO−
ROO− + H+ → ROOH
Bước này được đặc trưng bằng năng lượng chuyển electron – ETE.
PA= H(U−) + H(H+) – H(UH)
ETE= H(U•) + H (e−) – H(U−)
Enthalpy tổng của hệ ở điều kiện chuẩn (298K, 1atm):
Hf = E0 + ZPE + Htrans + Hrot + Hvib + RT.
1.2. Tổng quan các nghiên cứu về khả năng chống oxy hóa của các dẫn xuất từ urea
Nghiên cứu của Chamarthi Naga Raju và cộng sự (Năm 2017) đối với các dẫn xuất Urea/Thioure đi từ 3,5-dichloro-4-hydroxyl-aniline.
Nghiên cứu Kerr, tính toán, phân tích và tổng hợp được N-(2-(2-oxo-1-imidazolidinyl)ethyl)-3-phenylurea.
Eugene F. Hill và David O. De Pree đã tìm ra N-aryl- N’-(p-hydroxyphenyl) urea, dùng làm phụ gia chống oxy hoá cho xăng.
Năm 2016, Nhóm nghiên cứu của Sudhamani H. đã thiết kế và tổng hợp được nhiều hợp chất đi từ dẫn xuất của urea và thiourea, được ứng dụng trong thực tế để làm tác nhân chống oxy hoá và kháng khuẩn
Từ phenyl urea, Veera Raghavulu và cộng sự đã tính toán, thiết kế và tổng hợp được 1-[(1E)-2-(3-fluorophenyl) ethylidene]-3-(4-ethoxyphenyl) urea và 3-chloro-2-(3-fluorophenyl)-N-(4-methoxyphenyl)-4-oxoazetidine-1-carboxamide được ứng dụng trong y học, làm giảm quá trình lão hoá cơ thể người
Hợp chất có hoạt tính chất chống oxy hóa hiệu quả trong các sản phẩm dầu mỏ, nhựa, chất dẻo là các dẫn xuất của benzylurea. Cụ thể, Hotten và các cộng sự bằng đã đăng ký các bản quyền sáng chế về dãy các hợp chất mới dẫn xuất từ urea.
PhNCO + PhCH2NH2 🡪 PhNHCONHCH2Ph (Ph: nhóm phenyl: C6H5-) Các dẫn xuất bất đối xứng từ urea như PhNHCONHCH2Ph, PhNHCONHCH2Ph-3Me có hiệu quả chống oxy hóa rõ rệt.
Chương 2
LÝ THUYẾT LƯỢNG TỬ VÀ CÁC PHƯƠNG PHÁP
TÍNH TOÁN
2.1. Phương trình Schrödinger
Là phương trình cơ bản củ vật lý lượng tử, mô tả biến đổi trạng thái lượng tử của hệ vật lý theo thời gian.
Dạng đơn giản hoá:
Born−Oppenheimer đưa ra hàm sóng đầy đủ cho hệ n electron và m hạt nhân như sau:
2.2. Nguyên lý không phân biệt các hạt đồng nhất
Nguyên lý không phân biệt giữa các hạt đồng nhất chỉ ra “các hạt trong một hệ đồng nhất là không thể phân biệt được”.
2.3. Nguyên lý loại trừ pauli (nguyên lý phản đối xứng)
Nguyên lý loại trừ Pauli nói rằng không có hai electron (hoặc các fermion khác) có thể có trạng thái cơ học lượng tử giống hệt nhau trong cùng một nguyên tử hoặc phân tử.
Hàm sóng toàn phần mô tả trạng thái của hệ phải là hàm phản đối xứng.
2.4. Hệ nhiều electron
2.4.1. Hàm sóng
Ψel(x1,x2,…,xn)=χ1(x1).χ2(x2)… χn(xn)
Viết dưới dạng định thức Slater
2.4.2. Cấu hình Electron và trạng thái của hệ
Cấu hình electron bao gồm: Cấu hình vỏ đóng, cấu hình vỏ mở, cấu hình hạn chế, cấu hình không hạn chế.
2.5. Bộ hàm cơ sở
Có ba loại bộ hàm cơ sở: Bộ hàm tối thiểu, bộ hàm hóa trị, bộ hàm mở rộng.
2.6. Phương pháp tính toán
Một số phương pháp gần đúng với độ chính xác cao dùng để tính toán trong hoá học lượng tử:
2.6.1. Phương pháp Hartree−Fock
Phương pháp này được xây dựng dựa trên trường thế hiệu dụng trung bình đối với mỗi electron kết hợp với thế hút của hạt nhân và thế đẩy trung bình do các electron khác sinh ra. Phương trình hàm sóng theo phương pháp này được giải bằng phép lặp.
Phương pháp Hartree−Fock chỉ áp dụng cho hệ nguyên tử, khó áp dụng đối với hệ phân tử.
2.6.2. Phương pháp Roothaan
Khắc phục nhược điểm của phương pháp Hartree−Fock, tức là thay AO bằng MO.
2.6.3. Phương pháp bán kinh nghiệm
Phương pháp này thay thế một số tích phân trong phương trình hàm sóng bằng các tham số thực nghiệm để đơn giản hoá tính toán. Các phương pháp bán kinh nghiệm thường dùng là INDO, MINDO, AM1, NDDO, CNDO, PM3 …
2.6.4. Phương pháp nhiễu loạn
Lý thuyết này áp dụng cho 2 bài toán: Nhiễu loạn suy biến và nhiễu loạn không suy biến.
2.6.5. Phương pháp chùm tương tác (Coupled−Cluster−CC)
Phương pháp này xét đến cấu hình electron bị kích thích của hệ trong hàm sóng HF. Các phương pháp thường dùng CCSD (T) và CCSDT−1.
2.6.6. Thuyết phiếm hàm mật độ (Density Functional Theory−DFT)
Thuyết này biểu diễn hệ các electron bằng một hàm mật độ electron.
2.6.6.1. Cơ sở lý thuyết xây dựng DFT
Phương pháp DFT là sự kết hợp giữa dạng của phiếm hàm tương quan và phiếm hàm trao đổi và được giải bằng SCF (trường tự hợp).
2.6.6.2. Các phương pháp DFT thường dùng
2.6.7. Xây dựng bề mặt thế năng (pes – potential energy surface)
Bề mặt thế năng có thể mô tả thay đổi năng lượng toàn bộ của hệ
Xây dựng bề mặt thế năng để xác định toạ độ phản ứng nội, đó là điểm yên ngựa di chuyển xuống điểm cực tiểu.
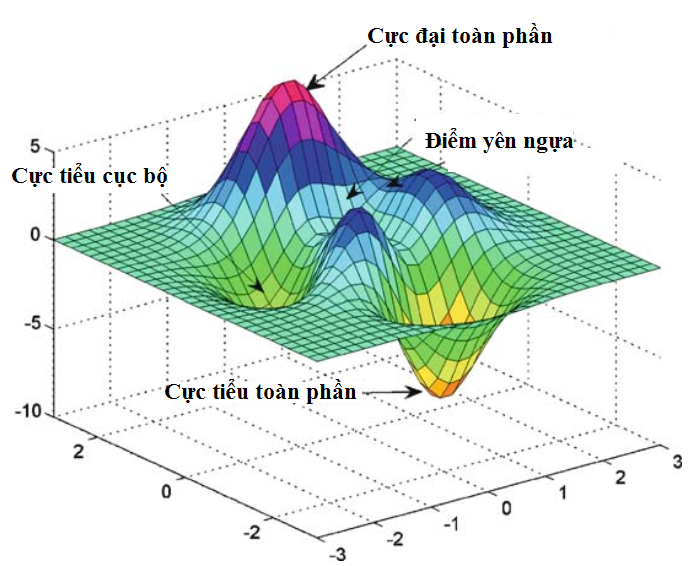
Hình 2.1. Bề mặt thế năng của phản ứng
2.7. Phương pháp tính toán áp dụng trong luận văn
2.7.1. Tối ưu hóa cấu trúc và tính năng lượng điểm đơn
Tối ưu hóa cấu trúc các chất với dòng lệnh tương ứng:
# B3LYP/6-311G(d,p) opt freq
Tính toán tại mức năng lượng lý thuyết cao hơn:
# ROB3LYP/6-311++G(2df,2p).
2.7.2. Tính toán các thông số nhiệt động học
2.7.2.1. Dữ liệu cơ bản nhận từ output file của gaussian 09
2.7.2.2. Tính toán các thông số nhiệt động liên quan
Năng lượng phân ly liên kết của liên kết: (BDE)
BDE(UH) = H(U•) + H(H•) – H(UH)
Năng lượng ion hóa của: (IE)
IE= Hf(UH+•) + Hf(e–) – H (UH)
Năng lượng phân ly proton (PDE)
PDE= Hf(UH•) + Hf(H+) – H(UH+•)
Ái lực proton (PA)
PA= H(U−) + H(H+) – H(UH)
Năng lượng chuyển electron (ETE)
ETE= H(U•) + H (e−) – H(U−)
Trong đó:
H(UH) là enthalpy của dẫn xuất urea
H(U•) là enthalpy của gốc tự do dẫn xuất urea
H(UH•+) là enthalpy của ion dương gốc tự do (radical cation) dẫn xuất urea
H(U−) là enthalpy của ion âm dẫn xuất urea
H(H•) là enthalpy của gốc tự do hydro
H(H+) là enthalpy của ion dương hydro
H(e-) là enthalpy của electron
Tổng enthalpy của cấu tử X, H(X), ở nhiệt độ T được tính bằng biểu thức sau:
H(X) = E0 + ZPE + Htrans + Hrot + Hvib + RT
Với: E0 là tổng năng lượng của hệ, ZPE là năng lượng dao động của hệ ở điểm không; Htrans, Hrot, Hvib tương ứng là enthalpy dịch chuyển tịnh tiến, enthalpy quay, enthalpy dao động trong hệ.
2.7.2.3. Tính toán và xây dựng bề mặt thế năng của phản ứng giữa chất nghiên cứu với gốc tự do
Bề mặt thế năng được tính toán và tối ưu cho từng chất phản ứng (reactants), sản phẩm (products) các phức có thể có (Int) và trạng thái chuyển tiếp (TS). Tối ưu trạng thái chuyển tiếp bằng dòng lệnh:
# opt(calcfc,ts) freq B3LYP/6-311G(d,p)
Số liệu thu được sẽ giúp tính toán năng lượng tương đối của các phức, trạng thái chuyển tiếp…Từ đó xây dựng được bề mặt thế năng.
2.7.3. Các phần mềm tính toán
Giới thiệu các phần mềm Gaussian 09W, phần mềm Gaussview 5, Eyringpy sử dụng trong hoá tính toán.
2.7.4. Sơ đồ quá trình tính toán
Chương 3
KẾT QUẢ VÀ THẢO LUẬN
3.1. Thiết kế và tối ưu hóa các dẫn xuất urea
3.1.1. Đánh giá sơ bộ hoạt tính chống oxy hoá của các dẫn xuất urea
Một số hợp chất urea được đề xuất nghiên cứu sơ bộ năng lượng phân ly liên kết N-H như Hình 3.1.
Hình 3.1. Tập hợp 10 dẫn xuất từ urea để đánh giá năng lượng phân ly liên kết
Các dẫn xuất urea được tính toán tối ưu cấu trúc ở dạng phân tử, dạng gốc tự do, sau đó xác định năng lượng liên kết và so sánh với số liệu thực nghiệm (nếu có).
Các liên kết trong vòng thơm benzene và nhóm CH3 tương đối bền (ví dụ BDE(C-H) = 112.9 kcal/mol cho benzene và 98.1 kcal/mol cho propane); do đó, hai loại liên kết dễ phân ly được quan tâm cho việc xác định năng lượng liên kết là N-H (vị trí số 1 và 3 trên Hình 3.1) và C-H (vị trí số 4 trong hợp chất U10). Các giá trị tính toán tại Bảng 3.1 nhằm hai mục đích a) đánh giá sơ bộ phương pháp tính toán đã đề xuất và b) xác định sơ bộ mức độ chuyển nguyên tử hydro đến gốc tự do để làm cơ sở cho việc thiết kế các dẫn xuất mới hiệu quả hơn.
Bảng 3.1. Năng lượng phân ly liên kết (BDE, kcal/mol) của 10 dẫn xuất urea (U) tính ở mức lý thuyết B3LYP/6-311G(d,p)
| Ureas | N1-H | N3-H | C4-H | Thực nghiệm | |
| U1 | 105.1 (107.8) | 105.1(107.8) | – | 111.0 | |
| U2 (CH3) | 103.6 (106.7) | 98.8(101.1) | |||
| U3 (di-CH3) | 98.7(101.3) | 98.7(101.3) | |||
| U4 (F) | 106.3(108.3) | 86.2(88.3) | |||
| U5 (di-F) | 87.4 (89.5) | 87.4 (89.5) | |||
| U6(F-CH3) | 84.6(86.8) | 99.4(101.4) | |||
| U7(Ph-H) | 101.7(104.3) | 89.5 (92.1) | |||
| U8(Ph-CH3) | 88.1(90.7) | 95.4(98.0) | |||
| U9(Ph-Ph) | 90.0(97.1) | 90.0(97.1) | |||
| U10 | 98.7(101.8) | 91.2(93.4) | 79.8(80.0) | ||
| Giá trị trong ngoặc đơn tính toán tại mức lý thuyết ROB3LYP/6-311++G(2df,2p)/ B3LYP/6-311G(d,p) | |||||
Kết quả tính toán cho thấy liên kết C−H dễ dàng bị cắt đứt và nguyên tử hydro dễ dàng chuyển đến kết hợp với gốc tự do, điều này sẽ đóng vai trò rất quan trọng trong phản ứng chống oxy hóa.
Như vậy, bên cạnh vai trò của liên kết N-H, cần quan tâm hơn các loại liên kết khác như C4-H trong hợp chất U10.
3.1.2. Thiết kế và tối ưu hóa các dẫn xuất urea mới có khả năng chống oxy hóa
Dựa trên các nghiên cứu trước đây, hai dẫn xuất mới U9a và U9b được đề xuất khảo sát.
Ngoài ra, dựa trên các kết quả nghiên cứu thực nghiệm, hợp chất mới là U10a cũng được đề xuất nghiên cứu.
Hình 3.2. Công thức cấu tạo của ba dẫn xuất urea từ U9 và U10
Kết quả tối ưu hóa cấu trúc của các chất nghiên cứu bao gồm chiều dài liên kết, góc liên kết và góc nhị diện được cho trên Bảng 3.2.
Bảng 3.2. Một số thông số cấu trúc lựa chọn của các chất nghiên cứu được tối ưu hóa bằng phương pháp B3LYP/6-311G(d,p)
| Thông số hình học cấu trúc | U9a | U9b | U10 | U10a |
| Chiều dài liên kết (Angstrom) | ||||
| N1-H | 1.008 | 1.008 | 1.009 | 1.010 |
| N1-C2 | 1.388 | 1.389 | 1.384 | 1.384 |
| C2=O | 1.216 | 1.214 | 1.218 | 1.220 |
| C2-N3 | 1.388 | 1.389 | 1.388 | 1.387 |
| N3-C | 1.410 | 1.406 | 1.408 | 1.408 |
| N1-C | 1.411 | 1.406 | 1.458 | 1.464 |
| N3-H | 1.008 | 1.008 | 1.008 | 1.008 |
| Góc liên kết (0) | ||||
| ∠N1C2N3 | 111.94 | 111.81 | 112.69 | 112.62 |
| ∠N1C2O | 124.03 | 124.09 | 122.81 | 123.07 |
| ∠C2N1C | 128.30 | 128.15 | 120.55 | 120.24 |
| ∠C2N3C | 128.30 | 128.15 | 128.47 | 128.59 |
| Góc nhị diện (0) | ||||
| Dih(CN1C2N3) | 179.94 | 179.76 | 174.19 | 173.81 |
| Dih(N1C2N3O) | 0.06 | 0.02 | 178.65 | 178.48 |
| Số liệu của U10 dùng để so sánh khi có nhóm thế CH3 tại meta | ||||
Kết quả trên cho thấy cấu trúc tối ưu nhận được bằng phương pháp B3LYP/6-311G(d,p) có các thông số hình học phù hợp với thực nghiệm; và trên cơ sở đó, có thể tính toán chính xác các thông số nhiệt động học liên quan khác.
3.2. Đánh giá khả năng chống oxy hóa của các dẫn xuất urea mới
Kết quả tính toán các thông số nhiệt động dựa trên cấu trúc hình học tối ưu của các dẫn xuất urea được trình bày trong Bảng 3.3.
Bảng 3.3. Năng lượng phân ly liên kết tính toán bằng phương pháp ROB3LYP/6-311++G(2df,2p)/ B3LYP/6-311G(d,p)
| BDE, (kcal/mol) | U9a | U9b | (U10) | (U10a) |
| N1-H | 86.9(N/A) | 90.6 (N/A) | 97.7(100.7) | 98.7(101.8) |
| N3-H | 86.9 (N/A) | 90.6 (N/A) | 91.2(100.2) | 91.2(93.4) |
| C-H | Không có | Không có | 79.9(80.3) | 79.8(80.0) |
| Giá trị trong dấu ngoặc đơn tính trong dung môi acetone Số liệu của U10 để so sánh khi có nhóm thế CH3 tại vị trí meta của vòng benzyl | ||||
So sánh các giá trị BDE(N-H) trong hợp chất U9a, U9b với U9 cho thấy nhóm đẩy điện tích CH3 và nhóm hút điện tử CF3 đều làm giảm BDE(N-H) trong U9a và U9b lần lượt khoảng 10 kcal/mol và 7 kcal/mol.
Kết quả cho thấy khả năng chuyển nguyên tử hydro tại vị trí liên kết C4-H dễ xảy ra hơn so với liên kết N-H.
Tuy nhiên, bên cạnh đó, yếu tố động học cũng đóng vai trò quan trọng trong việc xem xét khả năng chuyển hydro tại các liên kết khác nhau và sẽ được thảo luận chi tiết tại Mục 3.4.
Trên cơ sở đó, hợp chất U10 và U10a được đánh giá là hai dẫn xuất urea đại diện có hoạt tính chống oxy hóa; các thông số nhiệt động đặc trưng được tính toán như Bảng 3.4.
Bảng 3.4.Thông số nhiệt động tính toán trong pha khí tại ROB3LYP/6-311++G(2df,2p)
| Thông số nhiệt động | (U10) | (U10a) | ||||
| N1−H | N3−H | C4−H | N1−H | N3−H | C4−H | |
| IE | 175.7 | 174.8 | ||||
| PDE | 236.5 | 230.0 | 218.7 | 238.4(18.9) | 231.0(10.6) | 219.7(-2.8) |
| PA | 345.5 | 342.5 | 366.5 | 346.1(49.8) | 344.8(47.2) | 366.8(71.2) |
| ETE | 66.8 | 61.7 | 27.9 | 67.1(78.2) | 60.9(73.9) | 27.6(36.1) |
| Giá trị trong dấu ngoặc đơn tính trong dung môi acetone | ||||||
Nhìn chung, các thông số nhận được cho hai chất U10 và U10a không khác nhiều; Do đó, có thể kết luận rằng ảnh hưởng của nhóm CH3 tại vị trí meta trong hợp chất U10 là không đáng kể.
Dựa vào các phân tích và trên quan điểm nhiệt động học, cơ chế chuyển nguyên tử hydro (HAT) thuận lợi hơn trong cả pha khí và dung môi acetone.
3.3. Xây dựng bề mặt thế năng của phản ứng giữa một dẫn xuất urea tiêu biểu với CH3OO• và tính hằng số tốc độ phản ứng
3.3.1. Bề mặt thế năng của phản ứng giữa U10a với CH3OO•
Mô hình phản ứng của hợp chất U10a với CH3OO• được lựa chọn để tính toán bề mặt thế năng. Chất phản ứng, hợp chất trung gian, trạng thái chuyển tiếp và các sản phẩm được mô tả theo sơ đồ sau:
U10a-H + CH3OO• ⮀ Int1 (phức trung gian) ⮀ TS (trạng thái chuyển tiếp) ⮀ Int2 (phức sản phẩm) ⮀ U10a• + CH3OOH
Kết quả tính toán bề mặt thế năng của phản ứng tại các liên kết tương ứng được mô tả trên Hình 3.3
Kết quả cho thấy phản ứng chuyển nguyên tử hydro đến dập tắt gốc tự do tại C4-H thuận lợi hơn về mặt nhiệt động học. Tuy nhiên, chiều cao thế năng hoạt hóa khi chuyển nguyên tử H tại N3 lại thuận lợi hơn so với tạo vị trí C4(sp3); Do đó, để làm rõ hơn, các tính toán động học cho ba đường phản ứng sẽ được đề xuất tính toán ở Mục 3.3.2.
Đối với các đường phản ứng tại N3-H và C4-H, để làm rõ hơn sự phân bố electron, mật độ spin trong trạng thái chuyển tiếp, giá trị tính toán điện tích Mulliken và mật độ spin được trình bày trên Hình 3.4.
Hình 3.3. Bề mặt thế năng của phản ứng của U10a với CH3OO• tại phương pháp ROB3LYP/6-311++G(2df,2p)/ B3LYP/6-311G(d,p).
Giá trị điện tích tại nguyên tử H cho thấy proton H+ có xu hướng chuyển từ C hay N3 về phía O của CH3OO•, và khả năng phân ly H+ tại N3-H là mạnh hơn so với C-H. Điện tích H+ tại N3-H là +0.452, lớn hơn giá trị +0.289 tại C4-H là hoàn toàn hợp với lý do giá trị PA tại N3-H (344.8 kcal/mol) nhỏ hơn PA tại C-H (366.8 kcal/mol). Mặt khác, trạng thái TS tại N3-H thấp hơn so với tại C-H có thể được giải thích do mật độ spin tại N3 thấp hơn tại C4. Điều này có nghĩa là trong quá trình chuyển H, sẽ có sự giải toả mật độ electron nhiều hơn khi phản ứng xảy ra tại N3-H, làm cho rào năng lượng thế năng sẽ thấp hơn.
Tuy nhiên, một điểm cần nhấn mạnh là mặc dù trạng thái chuyển tiếp (TS) của đường phản ứng tại N3-H thấp hơn so với C4-H khoảng 2.2 kcal/mol, nhưng để xác định rõ hơn cơ chế phản ứng thì việc tính toán các hằng số tốc độ phản ứng là cần thiết.
4
4)
4
4
4
Hình 3.4. Phân bố điện tích Mulliken và mật độ spin (SD) trong trạng thái chuyển tiếp tại liên kết N3-H và C-H trong phản ứng U10a + CH3OO•
3.3.2. Động học phản ứng giữa U10a với CH3OO•
Sử dung phần mềm tính toán Eyringpy 1.02 cho từng đường phản phản ứng tại N1, N3 và C4. Trong đó, mỗi đường phản ứng gồm chất phản ứng (U10a và CH3OO•), phức chất hình thành khi CH3OO• tiến gần đến U10a (Int1), trạng thái chuyển tiếp (TS), phức chất sau khi nguyên tử H đã đến đầu O của CH3OO• (Int2) và sản phẩm phản ứng (U10a radical tại từng vị trí và CH3OOH).
Bảng 3.5 thể hiện năng lượng tự do Gibbs hoạt hóa và các giá trị tính toán hằng số tốc độ phản ứng tại 298.15 K cho mỗi đường phản ứng tại N1, N3 và C4.
Bảng 3.5. Năng lượng tự do hoạt hóa Gibbs (ΔG‡) và hằng số tốc độ phản ứng (k)
| Phản ứng chuyển H tại | ΔG‡ kcal/mol | k (cm3.molecule-1.s-1) | k ( L.mol-1.s-1) |
| N1-H | 18.6 | 3.0×10-21 | 1.81 |
| C4-H | 17.6 | 6.1×10-20 | 3.67×10 |
| N3-H | 14.3 | 1.3×10-17 | 7.83×103 |
Kết quả tính toán trên Bảng 3.5 cho thấy hằng số tốc độ phản ứng chuyển nguyên tử H đến gốc tự do CH3OO• được sắp xếp theo thứ tự N3 > C4 > N1. Điều này hoàn toàn phù hợp với bề mặt thế năng tính toán trên Hình 3.3. Tốc độ phản ứng tại N3 sẽ gấp 213 lần so với tại vị trí C4 và gấp 4330 lần so với vị trí N1.
Từ kết quả tính toán nhiệt động học và động học cho thấy yếu tố năng lượng phân ly liên kết là quan trọng để đánh giá khả năng dễ phân ly liên kết nhưng về phương diện động học thì cho thấy tại N3 tốc độ phản ứng lại xảy ra nhanh hơn. Do đó, tuỳ thuộc vào mục đích sử dụng, cụ thể tại nhiệt độ gia công các polymer hay nhiệt độ làm việc của các sản phẩm dầu mỏ, mà vai trò chuyển H tại N3 và C4 đều có sự đóng góp quan trọng trong quá trình chống oxy hóa hay chống sự suy giảm chất lượng sản phẩm. Ví dụ, tại 380 K (1070C), hằng số tốc độ phản ứng tại N3 chỉ còn lớn hơn tại C4 70.3 lần; ngoài ra, khi tăng nhiệt độ thì tốc độ phản ứng chuyển H tại C4 tăng nhanh hơn so với tốc độ phản ứng tại N3-H.
3.4. Đề xuất một số dẫn xuất Thiourea và Selenourea tương tự U10a
Với những kết quả nghiên cứu đã trình bày, có thể thiết kế một số dẫn xuất thiourea và selenourea tương tự như hợp chất (U10a) với mục đích tăng cường hoạt tính chống oxy hóa. Cụ thể có thể thay O trong nhóm carbonyl C=O bằng các nguyên tố S và Se, các hợp chất được ký hiệu U10a(S) và U10a(Se). Kết quả tính toán các thông số như trên Bảng 3.6.
Bảng 3.6. Các thông số nhiệt động của U10a(S) và U10a(Se) tính tại ROB3LYP/6-311++G(2df,2p) (đơn vị kcal/mol)
| Thông số (kcal/mol) | U10a(S) | U10a (Se) | ||||
| N1−H | N3−H | C4−H | N1−H | N3−H | C4−H | |
| BDE | 98.7 (101.8) | 91.2 (93.4) | 79.9 (80.0) | 90.4 (92.6) | 82.8 (86.9) | 79.5 (80.8) |
| IE | 164.6(105.1) | 160.2(100.6) | ||||
| PDE | 244.3 (20.4) | 235.3 (10.5) | 229.7 (2.5) | 244.7 (13.6) | 237.1 (19.3) | 233.8 (7.5) |
| PDE+IE | 408.9 (125.5) | 399.9 (115.6) | 394.3 (107.6) | 237.1 (114.2) | 404.9 (119.9) | 394.0 (108.1) |
| PA | 336.0 (41.4) | 335.2 (38.5) | 356.1 (63.3) | 332.8 (35.6) | 331.5 (39.0) | 353.2 (61.1) |
| ETE | 72.9 (84.0) | 64.6 (77.2) | 38.2 (44.3) | 72.1 (80.9) | 65.8 (78.6) | 40.8 (47.1) |
| PA+ ETE | 408.9 (125.5) | 399.9 (115.6) | 394.3 (107.6) | 237.1 (114.2) | 404.9 (119.9) | 394.0 (108.1) |
| Dữ liệu trong dấu ngoặc đơn được tính trong dung môi acetone. | ||||||
Phân tích cho thấy quá trình cho H tại C4-H là quan trọng trong các hợp chất nghiên cứu nên có thể nhận xét rằng khả năng chống oxy hóa theo HAT không bị ảnh hưởng nhiều bởi Se và S.
Dựa vào các số liệu có thể nhận xét rằng SETPT và SPLET ứng dụng phù hợp trong dung môi và cơ chế HAT đóng vai trò quan trọng trong pha khí.
3.5. So sánh khả năng chống oxy hoa của các dẫn xuất urea tiêu biểu với phenylenediamin
| Hợp chất | ZPE311 (kcal/mol) | TCE311 (kcal/mol) | TCG311 (kcal/mol) |
| U10a, C4-H | 77.9 | 79.6 | 78.9 |
| Phenylenediamin, N-H (Phụ gia trong xăng) | 120.8 | 124.6 | 115.7 |
| U10a(S), C4-H | 79.9 | – | – |
| U10a(Se), C4-H | 79.5 | – | – |
| ZPE311, TCE311 là giá trị BDE tính tại 0K và 298K và TCG311 là năng lượng tự do Gibbs | |||
Kết quả cho thấy các dẫn xuất urea được thiết kế có khả năng chống oxy hoá thuận lợi hơn nhiều so với phụ gia trong xăng (Phenylenediamin).
KẾT LUẬN VÀ KIẾN NGHỊ
- kết luận
Xây dựng được các cấu trúc hình học tối ưu cho các dẫn xuất từ urea với các thông số đáng tin cậy.
Đánh giá được năng lượng phân ly liên kết của các dẫn xuất urea.
Vai trò chuyển H của các liên kết N-H chưa thể hiện rõ rệt trong các hợp chất nghiên cứu.
Trong các phản ứng dập tắt gốc tự do bằng quá trình chuyển nguyên tử hydro, liên kết C4(sp3)-H của gốc benzyl (C6H5CH2-) và N3-H đóng vai trò quan trọng hơn so với liên kết N1H còn lại.
Cơ chế HAT được đánh giá ưu tiên hơn so với cơ chế SETPT và SPLET.
Trên cơ sở tính toán và xây dựng bề mặt thế năng phản ứng với CH3OO•, về mặt nhiệt động học, cho thấy phản ứng tỏa nhiệt tại C4-H và thu nhiệt tại N3-H và N1-H; tuy nhiên, về mặt động học phản ứng, tại vị trí N3-H, tốc độ phản ứng nhanh hơn tại C4-H 234 lần (298.15K). Yếu tố nhiệt độ ảnh hưởng lớn đến tốc độ chuyển H tại C4-H hơn tại N3-H.
Hợp chất U10a được đánh giá có khả năng chống oxy hóa theo cơ chế HAT. Trên cơ sở đó, đã đề xuất nghiên cứu hai hợp chất là U10a(S) và U10a(Se); kết quả cho thấy S và Se ảnh hưởng đến các giá trị nhiệt động học tính toán; tuy nhiên, với đại lượng BDE tại C4-H thì sự thay thế này là không đáng kể.
- Kiến nghị
Cần kiểm tra hoạt tính chống oxy hóa thông qua các kết quả thực nghiệm với DPP (Daily Practice Problems).
Tiếp tục tính toán và so sánh đặc tính chống oxy hoá của hợp chất nghiên cứu với các hợp chất chống oxy hoá đã được thương mại hoá khác để củng cố kết quả.
Xem xét định hướng tổng hợp để đưa vào ứng dụng trong thực tế.
E:\DỮ LIỆU COP CỦA CHỊ YẾN\DAI HOC DA NANG\KY THUAT HOA HOC\KHOA 35\(R)5. Pham Thanh Hai\TOM TAT



